龐貝氏症
」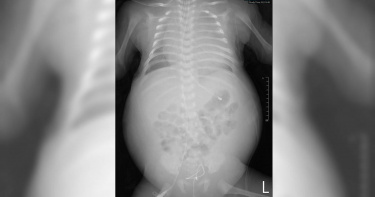
為保險延誤新生兒篩檢 27天大嬰兒險喪命
近一、二年來,約有2~3成新生兒父母,為了購買新生兒終生保險,在寶寶剛出生後只先接受法定的新生兒篩檢,約一至三個月左右核保通過後,再進行較全面的自費篩檢,這個觀念錯誤的情況,近幾年廣為流傳於家長社群中,但如無法即時篩檢出如龐貝氏症、脊髓性肌肉萎縮症(SMA)…等罕見疾病,可能錯失黃金治療時機,導致終生不可逆的傷害,甚至危及生命。台中一名出生27天的嬰兒王小弟,出現明顯的呼吸困難,緊急就醫後發現心臟異常肥大,肌張力低,肌肉酵素增高,高度懷疑為龐貝氏症,需插管治療。臺北榮總兒童醫學部楊佳鳳主任接獲通知後,立即派出醫療團隊,將病童接至臺北榮總,啟動緊急處置。雖到院當日立即完成診斷及給藥,但王小弟的病情仍然不樂觀。出現急性氣道壓迫,緊急進行「軟式支氣管鏡檢查」,立刻換置氣管內管,避免下端氣道完全壓迫無法進氣。在新生兒加護病房內,又頻繁出現心律不整,小小的胸腔被異常肥大的心臟塞滿,導致血液無法回流,合併全身水腫,心跳更降至每分鐘30下,情況十分危急,必須立即進行心臟按摩急救,新生兒葉克膜小組已在旁待命,經醫療團隊通力合作,成功挽救生命,住院治療40天,目前已康復出院多,未來僅需要每2周回來門診治療即可。龐貝氏症(Pompe disease)是一種罕見的遺傳性神經肌肉疾病,屬於體染色體隱性遺傳疾病。致病主要原因是溶小體一種酸性葡萄糖苷酵素(GAA)基因發生突變,導致GAA酵素無法分解肝醣,堆積的肝醣逐漸使肌肉受損,最後影響心臟和呼吸道肌肉。龐貝氏症疾病分成兩種類型,其中嬰兒型的患者若不及時治療,會在1歲前死於心臟衰竭或呼吸衰竭。在臺灣平均每2萬5千名新生兒就有1位是龐貝氏症患者。目前治療方式為每2周,施打病人體內所缺乏的葡萄糖苷酵素,越早治療預後越好。目前接受國內新生兒篩檢確診接受治療的約有100位病人。國民健康署補助21項新生兒篩檢,包括葡萄糖六磷酸鹽脫氫酶缺乏症G6PD(俗稱蠶豆症)…等等,詳如附表,惟不包含龐貝氏症、脊髓性肌肉萎縮症……等重大罕見疾病。王小弟的媽媽表示,「我們沒有做新生兒採檢,是因為剛出生的時候,我想要先為小孩規劃保險的部分,那等投保成功之後再去做自費篩檢,我沒想到會剛好,寶寶罹患龐貝氏症。」王媽媽表示,寶寶住院時甚至一度接到病危通知,幸好最後他挺過來了,真心呼籲大家「小朋友出生後盡早為小朋友做新生兒自費篩檢,這樣子小朋友跟爸爸媽媽,就比較不會需要承受驚慌和這麼煎熬的過程。」臺北榮總兒童醫學部牛道明主任呼籲每一位新生兒的家長,所有新生兒公費與自費篩檢項目,均經由專家學者反覆討論後制定,都是非常重要的疾病,尤其是龐貝氏症、脊髓性肌肉萎縮症(SMA)兩種罕見疾病,有著必須早期診斷、早期治療的急迫性。請各位家長務必依照正常程序篩檢,勿存僥倖心態,才不會延誤寶寶的治療,對長期預後造成重大影響。

9成於3歲前發病! 嬰兒「死亡率最高」遺傳罕病曝光
台灣新生兒連創新低,且每千位嬰兒就有4.1人死亡!台灣脊髓肌肉萎縮症病友協會理事長鐘育志表示,脊髓性肌肉萎縮症(SMA)為全世界嬰兒死亡率最高的遺傳疾病,患者有9成於3歲前發病,台灣患者死亡年齡中位數8.8個月,相較國外更嚴重。隨著衛福部本月起起擴大SMA用藥給付,醫界呼籲家長重視新生兒篩檢,及早發現問題以降低死亡率。每年篩出3千名異常寶寶 罕病八成為遺傳新生兒篩檢每年診斷出超過3千名異常個案數。然而,大多數遺傳性或先天異常疾病在寶寶出生時並無明顯症狀,但卻會在寶寶一出生時就造成傷害,若延遲篩檢或未篩檢,恐錯過黃金治療時機。台灣小兒神經醫學會秘書長郭雲鼎提醒,罕見疾病當中,高達八成為基因遺傳導致,想要找出潛藏疾病,需要透過新生兒篩檢來把關。目前衛福部國健署補助新生兒必選篩檢項目,包含先天性甲狀腺低能症、苯酮尿症、楓糖尿症、半乳糖血症等21個項目。郭雲鼎建議新手爸媽可以額外加選檢測約10項新生兒罕見疾病,如脊髓性肌肉萎縮症(SMA)、黏多醣症(MPS)、龐貝氏症(PD)等多種高帶因率的疾病。及早透過藥物或飲食可控制疾病,降低對身體或智力的損害,有更好的預後。SMA治療方式多 愈早篩檢與治療預後愈好鐘育志表示,在眾多新生兒遺傳性疾病中,脊髓性肌肉萎縮症(SMA)為全世界嬰兒死亡率最高的遺傳疾病,每10,000-17,181位新生兒中就約有1位會患病。患者有9成於3歲前發病,其中6成屬於重度的第一型患者,會在6個月大時發病。而且台灣患者死亡年齡中位數為8.8個月,比國外更嚴重。SMA屬於體染色體隱性遺傳疾病,脊髓的運動神經元會漸進性退化,產生肌肉無力、萎縮及行動困難等症狀。即使能存活下來,患者也會逐漸行動困難、進食與吞嚥困難、呼吸功能不全,不到20歲就得以輪椅代步,甚至逐漸凋零死亡。根據2023年「新生兒篩檢暨脊髓肌肉萎縮症(SMA)疾病認知調查」指出,家長不進行加選SMA篩檢的三大主因為:1不懂篩檢原因:四成因為沒有SMA相關家族病史、三成七因為不理解「新生兒篩檢加選SMA項目」內容而不進行篩檢2不懂嚴重程度:八成不知道SMA是世界上嬰兒死亡率最高的遺傳疾病3不懂有藥可醫:七成八不知道SMA確診後有藥物可治療鐘育志表示,現在SMA不再是無藥可治的疾病,若能及早篩檢、發現與治療,便能有更好的預後。目前台灣有三種治療藥物已通過藥證。健保署今年放寬SMA治療藥物給付範圍,期盼下一步能透過健保的幫助,讓不同族群的病患都能獲得急需的治療。

台灣嬰兒死亡率「高於日韓2倍」 3大主因錯失守護寶寶第一時機
台灣2022年新生兒數達62年來新低,不僅生得少,資料更顯示每千位嬰兒就有4.1位死亡,高出許多經濟合作暨發展組織(OECD)國家,更已連二十年超過日、韓2~3倍。台大醫院基因醫學部主任暨小兒部主治醫師簡穎秀部主任表示,寶寶健康狀況是家長最關心話題,但大多數遺傳性或先天異常疾病在寶寶出生時不會有明顯症狀,因此需要透過新生兒篩檢來把關,且所篩檢出來的疾病可透過藥物或飲食控制,及早降低疾病對身體或智力的損害,能有更好的預後。新生兒篩檢每年診斷出超過三千名異常個案數,但2023年「新生兒篩檢暨脊髓肌肉萎縮症(SMA)疾病認知調查」卻指出,不少家長為了守護寶寶的未來選擇犧牲現在。在願意讓寶寶進行加選篩檢家長中,超過95%家長會為寶寶保險,但卻近5成會延後篩檢時間,恐成寶寶健康的隱憂。調查中更發現,晚於常規時間才進行新生兒必選篩檢且為寶寶購買保險的家長中,卻有5成不清楚金融監督管理委員會在民國101年已頒布「新生兒篩檢之政府指定執行21項檢驗項目,皆排除於保險等待期的規範」。簡穎秀部主任提醒,別因商業保險迷思錯失拯救寶寶的機會!新生兒篩檢應在甚麼時間點進行?簡穎秀部主任表示,新生兒出生48小時即可篩檢,採取一次足跟血就可以進行必選及加選檢查。目前衛福部國健署有補助新生兒必選篩檢項目,包含先天性甲狀腺低能症、苯酮尿症、楓糖尿症、半乳糖血症等21個項目,也可以額外加選檢測約10項新生兒罕見疾病,為寶寶健康雙重把關。簡穎秀部主任說,家長一般會在生產後48小時內收到新生兒篩檢的勾選表,決策時間短,因此建議要提早了解。而篩檢時間也很重要,因為先天性代謝異常疾病一出生就會對寶寶造成傷害,及時篩檢有機會及早治療。但觀察願意進行篩檢的家長中,高達5成晚於常規時間才進行篩檢。進一步觀察原因,前三名分別為:決策進行篩檢時間太短、篩檢衛教資訊太少、太晚才開始了解篩檢項目,更有25%家長表示已進行產前帶因者檢驗或產前胎兒診斷,認為寶寶很健康。他補充,帶因檢驗並非100%準確,而產後新生兒篩檢更能直接確認寶寶的健康。以2022年出生率數字推估,高達一萬五家庭未進行加選篩檢!進一步觀察不進行「新生兒加選篩檢」原因,34%認為21項必選篩檢已足夠、近3成認為費用昂貴、近3成認為決策時間太短!台灣小兒神經醫學會秘書長、部立雙和醫院小兒神經科主任郭雲鼎秘書長提醒,罕見疾病當中,高達8成為基因遺傳導致。新生兒加選篩檢涵蓋多種可治療的罕病,如脊髓性肌肉萎縮症(SMA)、黏多醣症(MPS)、龐貝氏症(PD)等疾病檢測。以發生率高的脊髓性肌肉萎縮症(SMA)為例,篩檢準確度高。若延遲篩檢或未篩檢,恐錯過黃金治療時機。台灣脊髓肌肉萎縮症病友協會理事長、高醫大醫學院臨床醫學研究所講座教授鐘育志理事長補充,在新生兒眾多遺傳性疾病中,脊髓性肌肉萎縮症(SMA)為全世界嬰兒死亡率最高的遺傳疾病,每10,000-17,181位新生兒中就約有1位會患病。調查顯示,家長不進行加選SMA篩檢的三大主因為:1.不懂篩檢原因:4成因為沒有SMA相關家族病史、37%因為不理解「新生兒篩檢加選SMA項目」內容而不進行篩檢。2.不懂嚴重程度:8成不知道SMA是世界上嬰兒死亡率最高的遺傳疾病。3.不懂有藥可醫:78%不知道SMA確診後有藥物可治療。鐘育志理事長說,SMA屬於體染色體隱性遺傳疾病,6成患者會在6個月內發病,因負責製造運動神經元存活蛋白的基因SMN1發生突變,使脊髓的運動神經元漸進性退化,產生肌肉無力、萎縮及行動困難等症狀。現在SMA不再是無藥可治的疾病,若能及早篩檢、發現與治療,即有更好的預後。目前台灣有三種治療藥物已通過藥證。一種為脊髓腔內注射藥物,每四個月施打一次;一種為口服藥物,每日服用;以上兩種藥品機轉為能改變或修正SMN2基因的剪接,以增加完整的SMN蛋白。另一種基因治療,採用緩慢靜脈輸注一次性給予劑量,終身施打一次,機轉為將SMN1基因導入患者細胞內,代替缺陷基因的功能以生成完整的SMN蛋白。健保署今年將放寬SMA治療藥物給付範圍,期盼下一步能透過健保的幫助,讓不同族群的病患都能獲得急需的治療。

健保署今公布多項給付新制 放寬乳癌標靶藥給付範圍
健保署今年8月召開「全民健康保險藥物給付項目及支付標準共同擬訂會議(共擬會議)」,通過多項用藥提供病人臨床治療選用,包括「早期乳癌尚未發生腋下淋巴結轉移之病人可以用標靶藥物治療」、「遺傳疾病努南氏症可用生長激素治療」、「罕病龐貝氏症有更具安全的新藥可供選擇」、「類固醇治療無效之嚴重氣喘的12歲至17歲青少年,得以生物製劑治療」。健保署醫審及藥材組副組長張惠萍表示,本次共擬會議將乳癌標靶trastuzumab成分的藥品,放寬至「未發生腋下淋巴結轉移」也可給付,意即符合「HER2過度表現、雌激素受體(ER)為陰性、腫瘤大於2公分」等條件即可給付,估計每年能嘉惠510位乳癌病患。針對trastuzumab成分藥價,健保署也努力與廠商協商議價,張惠萍指出,本成分的原廠藥單價高達4萬3844元,若依現行最多給付六個月的方式估算,每人所花的健保費用為35萬元左右,但現在健保署已與兩家生物相似藥廠商議價為2萬9895元,每位病人所花的健保費僅23.9萬,成本已大幅減輕。不過此成分的給付範圍已擴大至未發生腋下淋巴結轉移,因此估計每年的健保赤字仍會達1億元左右。本次共擬會議還開放生物製劑治療嗜伊紅性白血球的嚴重氣喘患者,這項給付過去僅限於成人,張惠萍說,由於這款藥物已被證實在12歲以上病人使用也無虞,因此決議讓青少年在使用口服類固醇無效後,可由健保給付含mepolizumab成分藥品,預估能嘉惠163名青少年患者,每年將增加4400萬元赤字。但在罕病龐貝氏症的藥物使用上,每年最多可省下3.7億元,張惠萍表示,過去龐貝氏症病患僅能使用專案進口的alglucosidase alfa藥物,如今新藥avalglucosidase alfa已在國內取得藥證,因此成本已大幅縮減為每人每年一千萬,估計有97人受惠。此外,本次會議還通過遺傳性疾病努南氏症候群之兒童患者,可使用含somatropin成分之生長激素製劑,預估約64名兒童可脫離無藥可用的窘境。

中橡股東會通過0.1元現金股利 董座辜公怡:今年不確定性仍大
國際中橡(2104)今(7)日上午召開股東會,會中通過去年財報、配發0.1元現金股利,同時完成7席董監改選。董事長辜公怡表示,展望今年,上半年歐美國家逐漸走出疫情,市場供需回穩,但不確定性仍非常大,將審慎保守看待;生技事業龐貝氏症權利金爭議,最快年底將陸續有結果。針對碳黑事業,辜公怡指出,雖然中橡起步相對較晚,不過碳黑供需已逐步恢復穩定,目前台灣;中國、北美洲、印度等地共有8座廠,碳黑每年總產能79.5萬噸。至於碳黑新產能部分,印度新廠建設受疫情影響而延遲,明年上半年可望投產,同時也已在土耳其成立合資公司。因應防疫需求,中橡今年股東會現場採實名制入座,主管與股東分別至同棟樓不同會議室(圖/國際中橡提供)辜公怡強調,透過生產碳黑、回收製程中的蒸氣發電,1噸蒸氣可產生3戶人家1個月用電,這是中橡致力循環經濟的證明。未來中橡也將持續探索輪胎、橡膠製品、油墨塗料、導電產品等高質化產品的開發。針對生技事業部分,辜公怡指出,龐貝氏症孤兒藥權利金爭議案,歐洲,美國今年上半年均已完成聽審,預計美國今年底、歐洲明年上半年,將有進一步裁決。

陽光醫師驚罹肌萎症 扭轉命運幫助更多病友
艾爾莎(化名)高中時有一日感到腿部無力、背部疼痛,走路越來越慢,原以為是自己太累,但慢慢地、連站立都有困難,到處就醫的她,卻遲遲無法找出為何雙腳肌肉日漸萎縮的主因,直到某日出車禍送醫,醫師與護理師才警覺把她轉到神經內科,總算正式確診為肢帶型肌肉萎縮症。本身也是肌萎症病友的耕莘醫院病理科主治醫師、中華民國肌萎縮症病友協會理事長陳燕麟建議,若出現疑似肌肉無力症狀,應及早至神經內科求診,視情況進一步進行基因檢測,才能及早確診並選擇適合藥物或照護方式,目前部分罕見神經肌肉疾病,如龐貝氏症或部分類型肌萎症等皆已有治療方式,若能及早確診治療,或可有機會避免肌肉功能持續受損惡化、影響生活品質。陳燕麟醫師說明,罕見神經肌肉疾病雖然類型眾多,但在發病初期的症狀都很相似,例如肌萎症、龐貝氏症,都是因先天基因缺損或變異而導致肌肉細胞功能逐漸喪失,進而出現肌肉萎縮無力、起身或站立困難、走路越來越慢甚至走不動的情形,其中,肌萎症是一群多樣性肌肉退化性疾病的統稱,根據影響部位分成不同類型,目前國內盛行率約萬分之一,推估全台約有2300人罹病。陳燕麟醫師表示,除了症狀相似、不容易確診外,罕見神經肌肉疾病在發病初期的症狀較輕微,許多病友不易察覺或容易忽略疾病警訊,像是舉手越來越無力、雙腳無法跨大步伐,或坐下起身時常感到無力等,多數人可能誤以為是年紀大退化,或是腰椎壓迫、骨頭出問題,因此多選擇去推拿或到骨科、復健科就醫,以致於輾轉在各科診間數年,最後才順利被轉介到神經內科就診。罕見神經肌肉疾病初期症狀不易察覺,但若置之不理,一旦肌肉受損嚴重,將無法回復到原本正常功能,陳燕麟醫師建議,若出現起身困難、舉手無力、走路搖晃、容易跌倒等疑似肌肉無力的症狀,可以先做些平日較感吃力的動作自我檢測,例如快速蹲下起立或一次踩跨2、3階樓梯,若與同年紀者比較後發現肌肉功能較差,應及早至神經內科求診,視情況進一步接受基因檢測,才能及早確診並選擇適合藥物或照護方式,以減緩肌肉功能持續惡化。目前肌萎症已可透過基因檢測抽血來確診罹病類型,且過往檢體必須送到國外檢測,費用動輒高達十萬元以上,但隨著基因檢測技術精進與普及化,現已可在台灣檢驗,費用也降至約3萬至4萬元,雖然仍需自費負擔,但部分病友協會或組織有提供檢測補助,減輕患者及家屬的經濟壓力。病友艾爾莎在發現自己罹病後,經常主動分享自身經驗,希望能讓大眾與醫護人員更了解這個疾病,也幫助目前仍在各科診間輾轉流連的病友們,沒想到第一個受惠者竟是她的親弟弟,弟弟在高中首次發病後,艾爾莎立刻請家人帶弟弟到神經內科就醫,幸好很快就確診並得到很好的照護,省去輾轉流連各科的奔波辛苦。同是肢帶型肌萎症病友的陳燕麟醫師,原本是愛運動的陽光男孩,同樣經歷過輾轉各科才好不容易確診,卻發現這是個開刀也無法治癒的疾病時,他也曾久久無法平復心情,但後來選擇不跟命運妥協、轉換心境,開始鑽研罕見神經肌肉疾病,希望能透過病理科檢驗技術來幫助更多病友。一年多前接下中華民國肌萎縮症病友協會理事長一職後,更從過往在醫院被動等著病人上門求診走出去,偕同病友協會透過社工主動追蹤病友,提供醫療訊息、疾病照護、生活與情感上的支持。

鴻海捐贈12萬個有史以來最簡單的產品
鴻海科技集團今天宣布將捐出自產醫療口罩12萬片,給予包括家扶基金會等共10個社福團體,善盡企業社會責任。鴻海捐贈的口罩對象,除了家扶基金會外,還有罕見疾病基金會、原發性肺動脈高壓病友聯誼會、台灣泡泡龍病友協會、台灣龐貝氏症協會、尼曼匹克症病友聯誼會、紫質症病友聯誼會、淋巴血管平滑肌肉增生症(LAM)病友會、永齡慈善基金會、與台灣優質生命協會等全台灣10個社福團體。鴻海表示,今年2月初即於內部啟動口罩產線規劃,在不佔用社會資源下,包含相關機台設計、模具開發與組裝皆為自主完成,並在3月試產、申請認證,期間亦感謝經濟部工業局、衛福部食品藥物管理署、新北市政府、紡織研究所等單位給予指導、協助,後續配合政府徵用,一起攜手抗疫。在政府宣布採取「定量徵用」政策後,公司對於可自由調配的產能額度,即開始推動相關計畫,並決定捐贈12萬片口罩,協助經濟弱勢家庭、罕病朋友、以及獨居長者,讓他們不需要花費時間與金錢即有口罩可供防疫。鴻海透露,印有「Hon Hai/Foxconn」字樣的口罩,是一向以高精密製造為主產品,有史以來最簡單的一款,卻也是最有意義的。